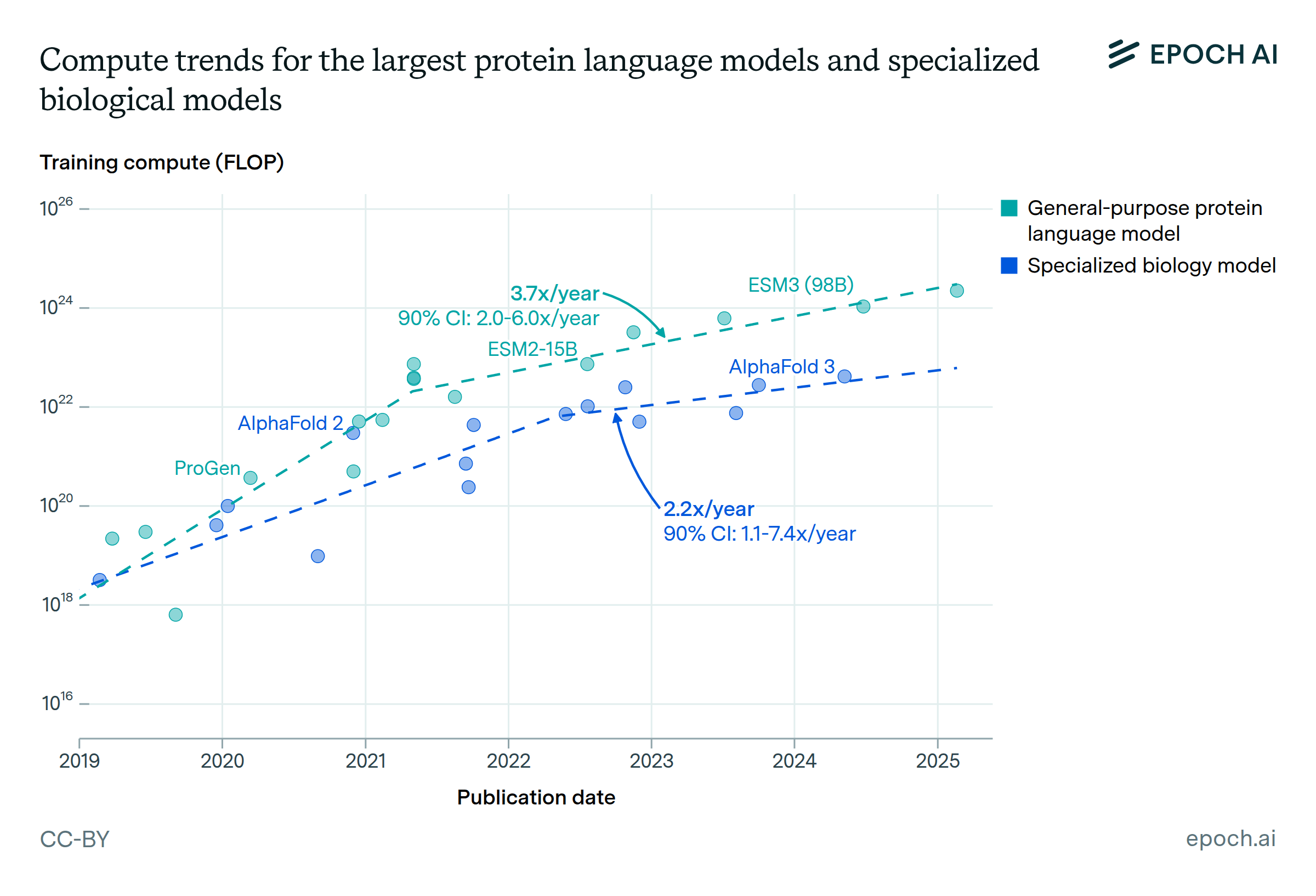
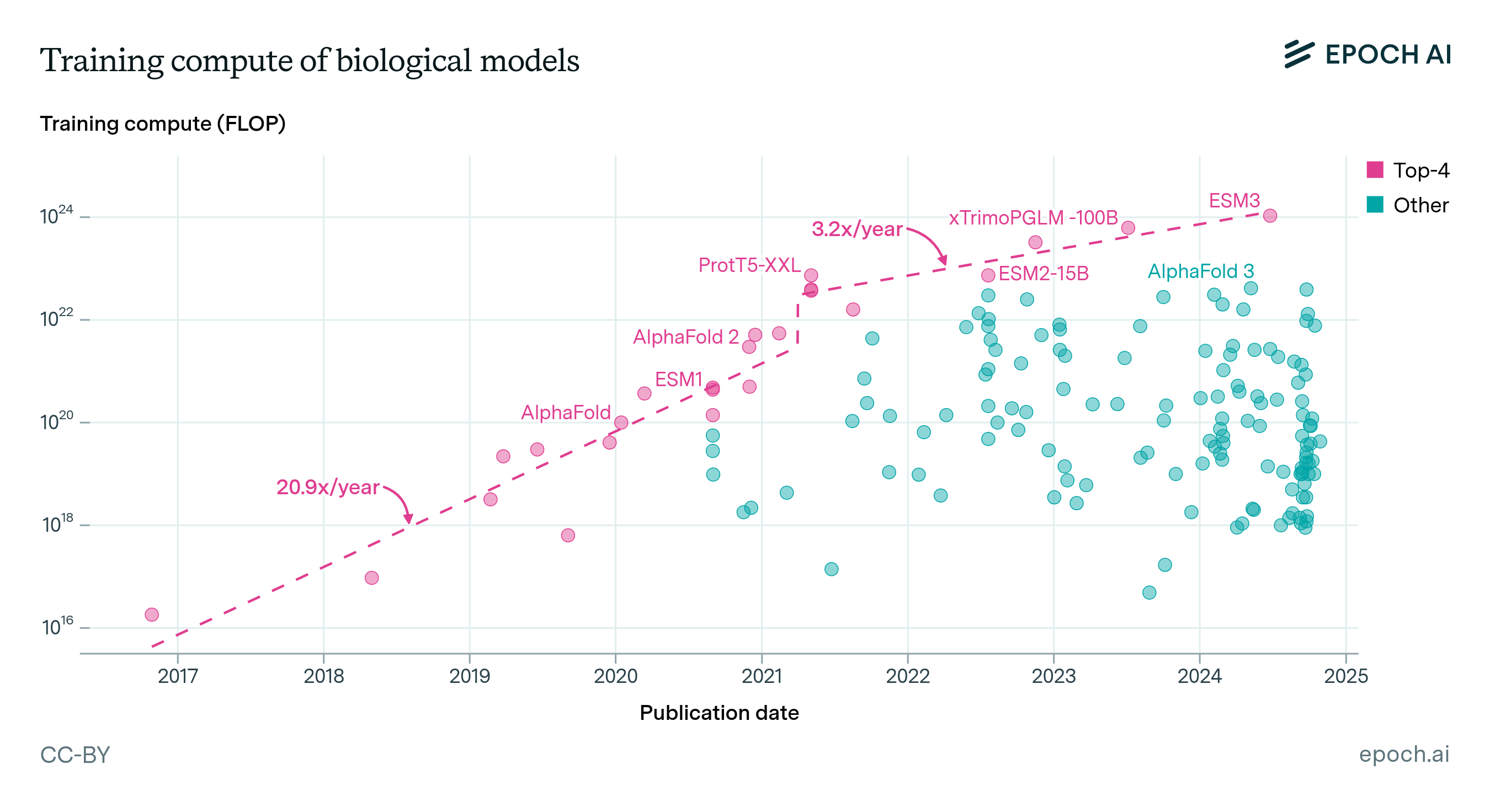
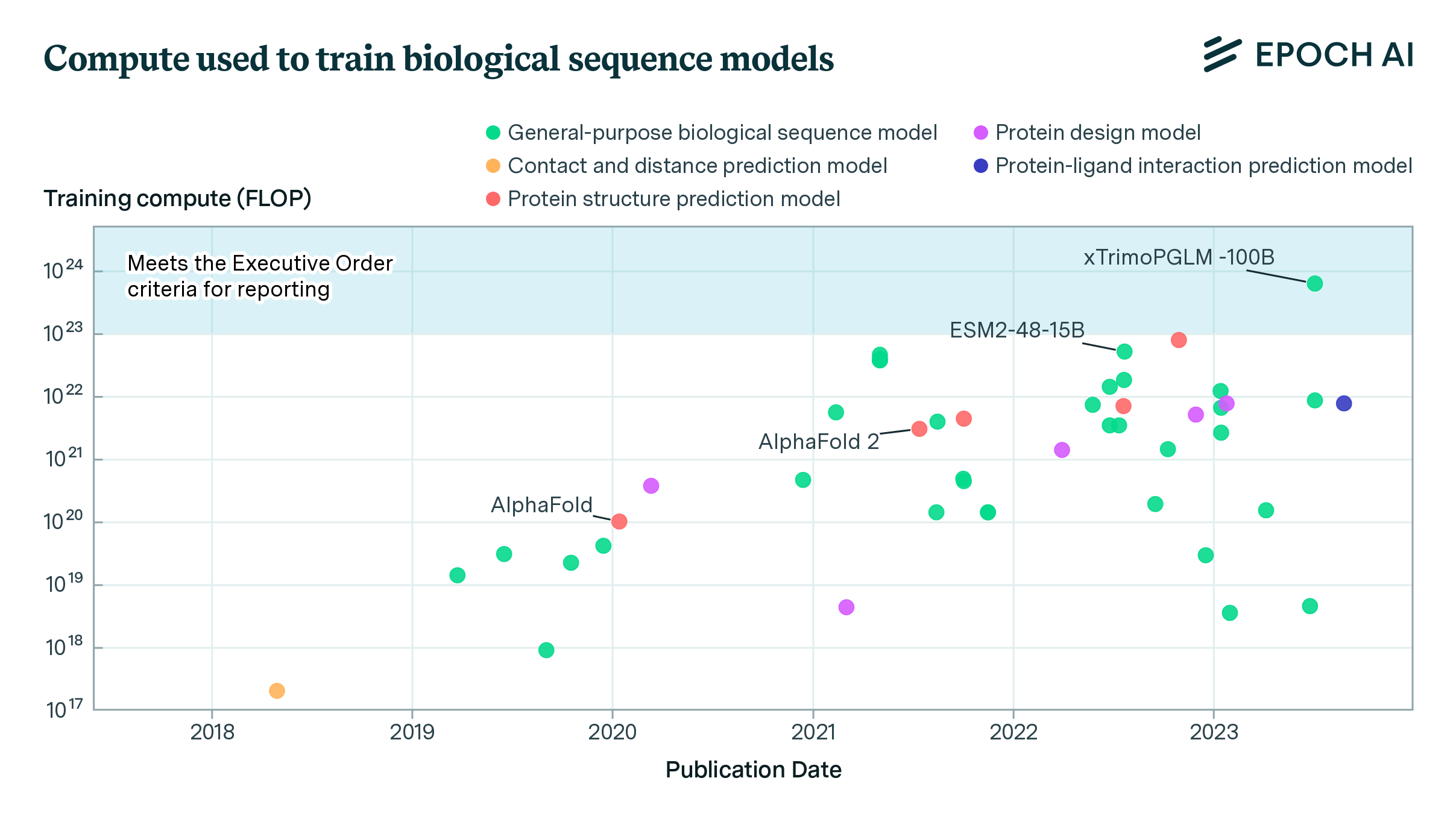
Expanding our analysis of biological AI models
We release a database of over 1,100 biological AI models across nine categories. We analyze their safeguards, accessibility, training data sources, and the foundation models they build on.
Published
This report presents an expanded database of AI models in biology, commissioned by Sentinel Bio and building on our 2024 collaboration, in which Sentinel Bio funded Epoch AI to collect and organize information about AI models in biology. The goal of this new report is to expand coverage to new categories of biology-relevant AI models and to capture releases since September 2024.
To build the database, we searched major academic databases and preprint servers for papers introducing AI models in biology, then used language models to filter candidates and extract structured metadata from the remaining papers. The most important models received additional manual review. The full methodology is described in the Appendix.
Key findings
The final database contains 1,196 models, of which 1,124 were annotated using AI assistance only while 72 received dedicated manual annotation. We also manually checked every entry for which we reported safeguards being used. Here are the main findings from our analysis:
Pre-release risk assessments and risk-related evaluations are rare. Only 2.5% of models have documented risk assessments, and 2.3% report running risk-related evaluations. These practices are more common among notable models, but this is largely driven by frontier LLMs, which account for 60% of the models in our database reporting risk assessments, and 68% of those reporting evaluations.
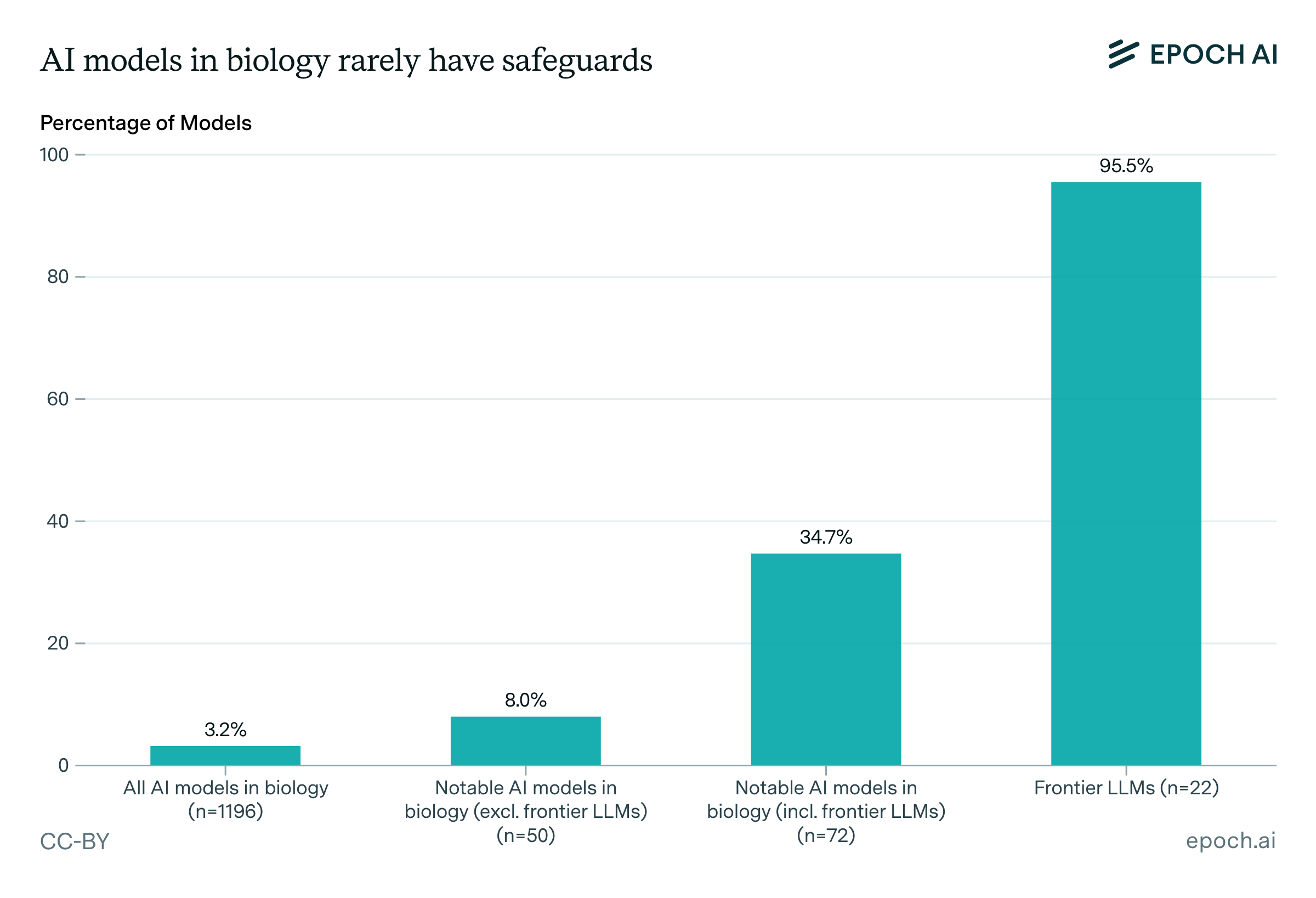
Safeguards are rare: only 3.2% of all models have any documented safeguards. Among notable models, 35% have safeguards, but again this is driven by frontier LLMs, 95% of which have safeguards, compared to just 1.4% of non-LLM biological AI models. Frontier LLMs account for over half of all models with safeguards.
When safeguards do exist, the types differ between frontier LLMs and other models. Frontier LLMs rely heavily on inference-time filtering, while non-LLM biological AI models with safeguards rarely use inference-time filtering, though they more commonly use post-training safety techniques or filter training data.
Most models share their code and data, though open weights are less common. About 58% of models provide open inference code and 46% share their training data. Open weights are released for about 23% of models.
Protein engineering and small biomolecule design are the largest model categories. These two model categories account for nearly half of the models in the database. Correspondingly, the most widely used training datasets are the Protein Data Bank and UniProt for protein-related, and ChEMBL for small molecule design models.
Many models build on a small number of foundation models. About one in five models in our database (253 of 1,196) are finetuned from an existing model rather than trained from scratch. The ESM-2 family of protein language models is by far the most common base.
Data access
The database containing the 1,196 models released after September 2024 is available at this link, with safeguard-related information redacted. Researchers and other parties with a legitimate interest in the safeguard data can request access by contacting data@epoch.ai.
Methodology overview
We aimed to capture AI models in biology1 released since September 2024, spanning nine categories: protein engineering, small biomolecule design, viral vector design, genetic modification, immune system modeling and vaccine design, pathogen property prediction, general-purpose biology AI, biology research agents, and biology desk research agents. We also included frontier large language models as part of the “general purpose” category.
Our process had four main stages. First, we searched for candidate papers across SemanticScholar, OpenAlex, and ChemRxiv using keyword and concept-based queries, yielding roughly 34,000 results. Second, we filtered these in two stages using AI-assisted screening: first on titles and abstracts, then on full paper texts. Third, we extracted structured metadata from the remaining papers (model name, category, parameters, training data, safeguards, accessibility, and other fields) using a prompted language model. Finally, we manually reviewed and annotated the 72 models we designated as “notable” based on criteria such as high citation counts, publication in top journals, or development by prominent research groups.
The full methodology, including search terms, extraction prompts, and detailed descriptions of each pipeline stage, is available in the Appendix.
Analysis
1. Dataset Overview
Our database contains 1,196 AI models in biology, of which 72 are designated as “notable” based on criteria including publication in prestigious journals, development by prominent research groups, or high citation counts. This section provides an overview of the landscape: where these models come from, who builds them, and what they do.
Category distribution
The distribution of AI models in biology across categories reveals the field’s current priorities. The two largest categories are protein engineering (e.g. given a target binding site, generate an amino acid sequence that folds into a protein that binds to it) and small biomolecule design (e.g. given a protein target, generate a small molecule that binds to it with high affinity). Together these constitute around half of the database. These, in addition to “Genetic modification” (13% of models), are the core “design” capabilities: tools for creating new proteins, drugs, and genetic sequences.

Pathogen property prediction (e.g. given a viral sequence, predict how transmissible or drug-resistant it is) is smaller but particularly relevant from a biosecurity perspective, given its dual-use potential. General-purpose models, a mixed category containing both frontier LLMs and biology-specific foundation models like ESM3 and Evo 2, account for about 13% of the database.
Geographic distribution
The geographic distribution of biological AI development is heavily concentrated. The United States and China together account for the majority of models, reflecting their dominant positions in both AI research and life sciences. A second tier of countries (the United Kingdom, Canada, Germany, and Switzerland) contribute meaningfully but at a much smaller scale. The remaining models are distributed across a long tail of countries, each contributing only a handful.
About 29% of models (249 of 865 with organization data) involve collaborations between organizations from multiple countries. This number likely underestimates the amount of international collaboration, since we track the country an organization is headquartered in, but many organizations have researchers located in multiple countries.

Institutional distribution
Universities produce the vast majority of publicly documented AI models in biology, followed by research institutes and corporations. This stands in contrast to frontier language model development, which is dominated by well-resourced industry labs. The pattern suggests that biological AI remains primarily an academic field, with much of the publicly accessible work driven by research groups rather than commercial entities. However, we have limited insight into biological AI models developed in-house at biotech and pharmaceutical companies.

Note that our organization data is incomplete: we link models to organizations through their associated papers, which does not always capture all contributing institutions, and geographic and institutional information is available for only about 72% of models. These figures should be treated as indicative of broad patterns rather than precise counts.
Notable models
Our database designates 72 models as “notable” based on several criteria: publication in prestigious journals, development by prominent research groups, or high citation counts. This subset allows us to examine patterns among the most impactful and visible models in the field.
The notable models subset reveals interesting differences from the full dataset. Corporations are far more prominent among notable models, a reversal of the pattern in the full dataset where universities dominate. This suggests that while academic labs produce the majority of AI models in biology, the most impactful and visible models tend to come from industry. The United States is also more preeminent among notable models compared to its share of all models.
The category distribution among notable models is dominated by “general-purpose” models, which includes both frontier LLMs and biology-specific foundation models like ESM3 and Evo 2.
2. Risk management practices
Some AI models in biology could potentially be misused to design or modify pathogens, toxins, or other harmful biological agents. Understanding how the field is addressing these risks is central to biosecurity policy discussions.
We track pre-release risk assessments as well as risk related evaluations. A pre-release risk assessment is a formal process conducted before deciding to release a model, such as threat modeling, documented risk analysis, or consultation with biosecurity experts. A pre-release risk-related evaluation is a test of whether the model can actually perform a risk-relevant task, for example by benchmarking its ability to generate pathogenic sequences or by conducting red-teaming exercises to probe for dangerous capabilities.
We also track which models implement safeguards to address biosecurity risks. Safeguards are measures taken by model developers to reduce potential harms. In the context of biological AI, relevant safeguards include filtering of harmful outputs, restrictions on who can access the model, and curation of training data to exclude dangerous sequences (such as those from human-infecting viruses).
The efficacy of safeguards is not assessed here and is beyond the scope of this analysis. Some safeguards can be easily removed. For instance, if data is purposefully excluded from pre-training, but the model weights are shared, the model can be fine tuned with task-specific data to recover the capability. For this reason, the mere presence of safeguards should not be interpreted as evidence that a model cannot be used for harm. There is ongoing work to develop and adapt a range of more robust safeguards to models of different types, but there may be technical limits to the extent to which open weights models can be secured.
Risk assessments and risk-related evaluations
Pre-release risk assessments and risk-related evaluations are rare. Only 2.5% of models have documented risk assessments, and 2.3% report running risk-related evaluations, and 3% of models report running at least one of the two. These practices are more common among notable models, but this is largely driven by frontier LLMs, which account for 60% of the models in our database reporting risk assessments, and 68% of those reporting evaluations.

Among frontier LLMs, pre-release risk management is now standard practice. Models like GPT-5.2, Claude Opus 4.6, and Gemini 3.0 Pro all report conducting both risk assessments and evaluations for dangerous biological capabilities before release. Outside of frontier LLMs, a smaller number of biology-specific models have adopted similar practices, including Evo 2 (a genomic foundation model from Arc Institute) and Google’s AI Co-Scientist.
Safeguards
Safeguards are rare: only 3.2% of all models have any documented safeguards. Among notable models, 35% have safeguards, but again this is driven by frontier LLMs, 95% of which have safeguards compared to just 1.4% of other models. Frontier LLMs account for over half of all models with safeguards.
When safeguards do exist, the types differ between frontier LLMs and biology-specific models. Frontier LLMs rely heavily on inference-time filtering (present for 95% of Frontier LLMs with safeguards) or access restrictions (62%), while biology-specific models with safeguards rarely use inference-time filtering (6%). Instead, biology-specific models more commonly use post-training safety techniques (65%) or filter training data to exclude dangerous sequences (35%). For example, Evo 2 and the open-weights version of ESM3 filter training data to exclude certain viral and other pathogen-related sequences.

3. Accessibility
Accessibility refers to how openly model developers share their work. We track several dimensions, which are not mutually exclusive (e.g. a model can be open weights but not open data, or vice versa):
- Open inference code: The code needed to run the model is publicly available.
- Open training code: The code used to train the model is shared.
- Open weights: The model’s learned parameters are released, allowing others to use the model without retraining.
- Open data: The training dataset is available.
- API access: The model can be queried through a programming interface, typically with some access controls.
- Web interface: The model is accessible through a browser-based tool.
- Unreleased: The model has been described in a publication but is not publicly available.

Most models do not restrict access. About 58% of models provide open inference code, and 46% share their training data. Open weights (23%) and open training code (28%) are less common but still substantial. Only a small fraction of models are available only through API access (3.8%).
4. Building Block Models
Many AI models in biology are not trained from scratch but are instead “finetuned” from existing foundation models. Finetuning involves taking a pre-trained model and adapting it to a specific task using a smaller, specialized dataset. This approach is efficient because the base model has already learned general patterns (like protein structure or sequence relationships) that transfer to downstream tasks.
In biology, common finetuning tasks include predicting protein function, identifying binding sites, classifying sequences by organism or function, or predicting the effects of mutations. The base models are typically large language models trained on biological sequences (like protein or DNA sequences) that have learned rich representations of biological structure and function.

Among the 253 models in our database that are finetuned from another model, ESM-2 is by far the most common base model, with 58 models built on top of it (this count aggregates across ESM-2 versions of different sizes). ESM-2 is a protein language model trained on millions of protein sequences.
Other common base models include ProtBERT (15 models), AlphaFold2 (7 models), and ProtGPT2 (7 models). The prominence of these protein-focused models reflects the field’s emphasis on protein engineering and design.
5. Training Data, Parameters, and Compute
Training datasets
Among the 1,009 models with training dataset information, the Protein Data Bank (PDB) is the most widely used, appearing in 159 models. UniProt is second (93 models), followed by ChEMBL (69 models) which is most useful for small molecule and drug design. GISAID (38 models), which comes next, contains influenza and coronavirus sequences and is primarily used for pathogen-focused models.

Protein and small molecule datasets account for the bulk of reported training data usage: this is consistent with those categories having the most models. Genomic databases are overall less prominent.
Parameters, data size, and compute
This report differs from typical Epoch AI outputs on frontier models, where training compute is often the central focus. For biological AI, we do not emphasize compute as heavily. The long tail of model architectures makes compute estimation much harder than for language models, where a few well-understood architectures cover most of the field. But more importantly, compute does not appear to be the primary bottleneck for biological AI capabilities (and might be even less relevant when considering capabilities related to misuse risks); progress seems more constrained by data availability and quality than by raw compute. That said, some observations are worth noting:
Parameters: Biological AI models span many orders of magnitude in size. Most models in our database are relatively small by modern standards, ranging from tens of millions to a few billion parameters. The largest biology-specific models, like Evo 2 (40 billion parameters) and xTrimo’s 100 billion parameter model, approach the scale of medium-sized language models but remain smaller than frontier systems like GPT-5 or Claude.
Compute: Training compute estimates are available for only a small subset of models. Where reported, values range from 1017 to 1023 FLOP, roughly spanning from a single GPU-day to a small fraction of frontier language model training runs (in the high hundreds of thousands of dollars for a 1023 FLOP run). The largest biology-specific training runs remain 2-3 orders of magnitude smaller than frontier AI training.
Appendix
Appendix A: Detailed methodology
Scope and inclusion criteria
We aimed to capture AI models in biology released around or after September 2024, spanning nine categories adapted from RAND Europe and the Center for Long-Term Resilience’s Global Risk Index for AI-enabled Biological Tools:
- Protein engineering tools (design, prediction, and analysis of amino acid sequences and protein structures)
- Small biomolecule design tools (optimization and de novo design of small molecules and peptides)
- Viral vector design tools
- Genetic modification and genome design tools
- Immune system modeling and vaccine design tools
- Pathogen property prediction and host-pathogen interaction tools
- General-purpose AI models trained on significant quantities of biological data, especially biology AI foundation models
- Biology research agents that can make use of the above tools
- Biology desk research agents for ideation and planning in these domains (e.g. via literature review)
We also included frontier large language models where relevant to categories 7 and/or 9.
Search
The goal of the search stage was to assemble a broad list of candidate papers that might describe the training of a biology-related AI model in one of our categories. We aimed for complete coverage of notable models (goal 1) and broad coverage of the long tail of less prominent but still relevant models (goal 2). Notable models known to us were added manually to satisfy goal 1; the automated search process described below addressed goal 2.
We searched three sources:
- SemanticScholar. We used the SemanticScholar search API with eight sets of keyword queries, one per category. The initial keyword sets were provided by collaborators from RAND Europe and the Center for Long-Term Resilience and then modified to work with the SemanticScholar API’s syntax and to improve recall. The full search terms are listed in Appendix C.
- OpenAlex. We supplemented the SemanticScholar results with both a keyword-based search and a concept-based search on OpenAlex, which surfaced additional papers not returned by SemanticScholar.
- ChemRxiv. We also searched ChemRxiv to capture preprints in chemistry and related fields.
Results from all three sources were combined and deduplicated. All metadata returned by the APIs (titles, abstracts, authors, publication dates, etc.) was stored and used in later pipeline stages.
Filtration
The search process yielded roughly 34,000 candidate papers. The filtration stage aimed to exclude papers that did not describe the training of a model fitting one of our categories.
Abstract-level filtering. The first pass used GPT-5-mini, prompted with each paper’s title and abstract. The model was briefed on the types of papers to include and given explicit examples of papers that should be excluded. It responded with “Yes,” “No,” or “Maybe” for each paper. We included “Maybe” papers in the next stage because at this threshold, the cost of missing a relevant paper exceeded the cost of screening additional full texts.
Full-text filtering. Papers that passed abstract screening were then filtered using the full text of the paper in PDF format. The model was prompted with a similar goal as the abstract stage but with access to the complete paper.
Paper full-text retrieval
Only papers that passed abstract screening needed their PDFs for full-text filtering and information extraction. We automatically retrieved papers from the arXiv and bioRxiv S3 buckets where available, which covered a large portion of the corpus. Of the 643 papers that could not be retrieved automatically, 478 were downloaded manually (those available without purchasing individual access). The remainder, which required individual purchase, were excluded from the analysis.
Information extraction
For each paper that passed full-text filtering, we used Gemini 3.0 Flash (high thinking setting) to extract structured metadata from the PDF. The model was prompted to identify each distinct AI model described in the paper and extract fields including model name, category, parameters, training data, safeguards, accessibility, and finetuning relationships. The full extraction prompts are available in Appendix D.
We ran separate extraction passes for the general metadata, safeguard information, and category tags. We iterated over the prompts over multiple runs before choosing the final prompts.
The extracted information was organized into two tables: Systems (the AI models) and Papers (the source publications). The full schema for these tables is described in Appendix B.
The choice of fields was motivated by two goals: understanding the safeguards and safety-relevant features of these models, and capturing the variety of relationships between models (finetuning) along with other metadata (release date, papers, authors, training datasets) that consumers of the database might find useful.
Notability criteria and manual annotation
Some models warrant more careful documentation than others. We designated models as “notable” if they met any of the following criteria:
- Created by a group or person noteworthy in biology, AI, or a related field
- Associated with a GitHub repository with more than 1,000 stars (a proxy for uptake and public interest)
- Associated with a paper cited over 100 times
- Published in Nature or Science
- Notable by our discretion for reasons not captured by the above
Notable models received dedicated manual annotation: a researcher reviewed the paper and verified or corrected all extracted fields.
Appendix B: Database Fields
The database consists of two tables. The columns for each are listed below.
Systems table
| Column | Column description |
|---|---|
| Name | Name of the model |
| Primary category | The single best-fit category |
| All categories | Every category the model could reasonably fall into - this field aggregates the two categories “Research agent” and “Desk research agent” |
| Is frontier LLM | Whether the model is a frontier LLM like GPT-5, Claude Sonnet 4, etc. |
| Accessibility | Tags describing model availability (e.g. open weights, API access) |
| Accessibility notes | Notes on model accessibility |
| Release date | Date of release, typically approximated by publication date |
| Paper | Link to the corresponding Papers entry |
| Notability criteria | For notable models, which criteria were met |
| Training datasets | Link to the corresponding Datasets entries |
| Training datasets notes | Detailed description of the model’s training data |
| Tokens trained on | Number (sometimes estimated) of tokens trained on in training |
| Is a finetune of | If a finetune, link to the base model |
| Is the base model for | Reciprocal of “Is a finetune of” |
| Implements | For agents, links to models used via inference |
| Implemented by | Reciprocal of “Implements” |
| Training compute (FLOPs) | Estimated training compute in floating-point operations |
| Training compute notes | Notes on the compute estimate |
| Parameters | Number of model parameters |
| Parameters notes | Notes on the parameter count |
| Attention received | Level of human review this model received during annotation |
Papers table
| Column | Column description |
|---|---|
| Title | Title of the paper |
| Models | Links to extracted Systems entries |
| Link | URL to the paper |
| Authors | Authors of the paper |
| Abstract | Abstract of the paper |
| Published | Publication date |
| Citations | Citation count as of February 2, 2026 |
| Organizations | Affiliated organizations |
| Attention received | Level of human review during annotation |
| Added by | Whether the paper was added manually or surfaced by the automated pipeline |
Appendix C: Search terms
SemanticScholar search terms
"Protein engineering": "(\"protein design\" | \"protein folding\" | \"protein inverse folding\" | \"protein diffusion model\" | \"de novo design\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Small biomolecule design": "(\"biomolecule design\" | \"ligand structure prediction\" | \"molecular structure prediction\" | \"de novo design\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Viral vector design": "(\"viral vector\" | \"capsid design\" | \"capsid engineering\" | \"vector engineering\" | \"viral delivery system\" | \"viral vector optimization\" | \"AAV\" | \"adeno-associated virus\") (\"machine learning\" | \"artificial intelligence\" | \"computational model\" | \"AI\" | \"deep learning\" | \"neural network\")",
"Genome modification and genome design": "(\"genome assembly\" | \"genomic assembly\" | \"sequence assembly\" | \"DNA assembly\" | \"genome reconstruction\" | \"sequence reconstruction\" | \"contiguous assembly\" | \"contig assembly\" | \"genetic feature identification\" | \"gene annotation\" | \"motif discovery\" | \"regulatory element identification\" | \"regulatory factor binding identification\" | \"gene regulatory element mapping\" | \"cis-regulatory element analysis\" | \"codon sequence optimisation\" | \"codon sequence optimization\" | \"codon optimisation\" | \"codon optimization\" | \"codon bias adjustment\" | \"promoter sequence optimisation\" | \"promoter sequence optimization\" | \"genome design\" | \"genome synthesis\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Pathogen property prediction": "((\"viral\" | \"virus\" | \"bacteria\" | \"bacterial\" | \"pathogen\") (\"prediction\" | \"predict\" | \"detection\" | \"detect\") (\"zoonotic spillover\" | \"host tropism\" | \"environmental stability\" | \"transmissibility\" | \"virulence prediction\") | (\"toxicity prediction\" | \"toxic molecule prediction\" | \"toxic peptide prediction\")) (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Host-pathogen interaction prediction": "((\"virus\" | \"bacteria\" | \"pathogen\" | \"host\") (\"viral host entry\" | \"virus host entry\" | \"immune escape\" | \"antibody escape prediction\" | \"antibody escape predict\") | (\"protein\u2013protein interaction prediction\" | \"protein-protein interaction prediction\" | \"protein\u2013protein interaction predict\" | \"protein-protein interaction predict\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Immunological system modeling and vaccine design": "(\"immune system model\" | \"immunological system model\" | \"immune response predict\" | \"immune response detect\" | \"immune response prediction\" | \"immune response detection\" | \"t-cell receptor epitope predict\" | \"t-cell epitope detect\" | \"t-cell epitope prediction\" | \"t-cell epitope detection\" | \"clinical outcome predict\" | \"clinical outcome detect\" | \"clinical outcome prediction\" | \"clinical outcome detection\" | \"immune model\" | \"immunological model\" | \"immunogenicity prediction\" | \"immunogenicity predict\" | \"immunogenicity detection\" | \"immunogenicity detect\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")|(\"vaccine design\" | \"vaccine develop\" | \"vaccine engineer\" | \"vaccine subunit design\" | \"vaccine subunit prediction\" | \"vaccine subunit predict\" | \"antigen prediction\" | \"antigen predict\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\" | \"automat\")",
"Experimental design, simulation and automation": "((\"experimental simulation\" | \"experiment simulation\" | \"in silico experiment\") (\"biotechnology\" | \"bioinformatics\" | \"synthetic biology\" | \"computational biology\" | \"genomics\" | \"proteomics\" | \"biological tools\" | \"bio-engineering\" | \"biomedical engineering\" | \"bioprocessing\" | \"biomanufacturing\") | (\"experiment design\" | \"experimental design\" | \"experiment method\") (\"bio\" | \"clinical\" | \"medicine\" | \"pharma\")) (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\")|(\"automated\" | \"automate\" | \"automatic\" | \"autonomous\") (\"experiment\" | \"cloud lab\") (\"bio\") (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"neural network\" | \"computational model\")",
"Literature analysis and research agents": "((\"literature review\" | \"systematic review\" | \"literature synthesis\" | \"literature mining\") (\"automated\" | \"automate\" | \"automatic\" | \"AI-assisted\" | \"AI-powered\") (\"biological\" | \"biomedical\" | \"genomic\" | \"molecular biology\" | \"biotechnology\") | (\"research agent\" | \"AI agent\" | \"autonomous agent\") (\"literature\" | \"scientific literature\") (\"biological\" | \"biomedical\" | \"life science\")) (\"machine learning\" | \"artificial intelligence\" | \"deep learning\" | \"large language model\" | \"LLM\")"
}
Appendix D: Prompts
Metadata extraction
You are provided with a research paper (attached PDF) that has ALREADY passed the relevance gate.
Your task: extract metadata for ALL in-scope biology AI models described in the paper.
=========================================================================
EXTRACTION SCOPE
=========================================================================
Extract data for EVERY distinct AI model or agent that the paper introduces, releases, or substantially fine-tunes within this scope:
1. Protein engineering tools
2. Small biomolecule design tools
3. Viral vector design tools
4. Genetic modification and genome design tools
5. Immune system modelling and vaccine design tools
6. Pathogen property prediction and host-pathogen interaction tools
7. General-purpose biology AI
8. Biology research agents / desk research agents
If the paper describes multiple model variants (e.g., "ModelX-Small", "ModelX-Large"), extract each as a separate entry.
=========================================================================
CATEGORY DEFINITIONS
=========================================================================
Each model must be assigned to EXACTLY ONE category (the best fit).
Below are detailed definitions. Use these to guide your classification:
---
**1. "Protein engineering tools"**
Scope: AI systems for the design, optimization, prediction, or analysis of amino acid sequences and protein structures, when the purpose is to enable or drive sequence/structure engineering.
Includes:
- De novo protein design (e.g., generative models for novel proteins)
- Protein structure prediction models (e.g., AlphaFold-like models)
- Protein function prediction for engineering purposes
- Antibody design and optimization
- Enzyme engineering and optimization
- Protein-protein interaction prediction for design
- Protein fitness landscape modeling
- Directed evolution guidance models
Does NOT include: Models that only interpret experimental protein data (e.g., mass spec analysis) without a design component.
---
**2. "Small biomolecule design tools"**
Scope: AI systems for the optimization or de novo design of small molecules and peptides, especially for drug discovery or metabolite design.
Includes:
- Molecular generation models (VAEs, diffusion, autoregressive)
- Molecular property prediction for optimization
- Drug-target interaction prediction
- Lead optimization models
- Retrosynthesis planning
- Peptide design (short sequences, typically <50 amino acids)
- ADMET property prediction when used for design
Does NOT include: Simple QSAR models used only for screening without generative capability.
---
**3. "Viral vector design tools"**
Scope: AI systems that design or optimize viral vectors and capsids for gene therapy, delivery, or related applications.
Includes:
- AAV capsid design and optimization
- Viral vector tropism prediction
- Capsid engineering for improved delivery
- Lentiviral vector optimization
---
**4. "Genetic modification and genome design tools"**
Scope: AI systems that propose or optimize DNA/RNA sequences, edits, or larger genome designs.
Includes:
- CRISPR guide RNA design and optimization
- Gene circuit design
- Regulatory element (promoter/enhancer) design
- Codon optimization
- Synthetic genome design
- Operon and construct design
- Gene expression prediction for design purposes
- Variant effect prediction when used to guide editing
Does NOT include: Models that only interpret sequencing data without design intent.
---
**5. "Immune system modelling and vaccine design tools"**
Scope: AI systems for designing vaccines, immunogens, epitopes, or modeling immune responses to guide design.
Includes:
- Epitope prediction and design
- Immunogen optimization
- Vaccine antigen design
- Antibody repertoire modeling
- MHC binding prediction for vaccine design
- Neoantigen prediction for cancer vaccines
---
**6. "Pathogen property prediction and host-pathogen interaction tools"**
Scope: AI systems modeling pathogen traits or host-pathogen interactions, particularly for biosecurity-relevant applications.
Includes:
- Pathogen virulence prediction
- Transmissibility prediction
- Drug resistance prediction
- Immune escape / antigenic drift prediction
- Zoonotic risk assessment
- Pandemic potential modeling
- Host-pathogen protein interaction prediction
IMPORTANT: This is a HIGH-PRIORITY category for biosecurity tracking. Do not miss models in this category.
---
**7. "General-purpose biology AI"**
Scope: Large foundation models trained on substantial biological data that support multiple downstream tasks, or are explicitly described as "foundation", "general", or "platform" models.
Includes:
- Multi-modal biological foundation models (e.g., protein + DNA + molecules)
- Large-scale biological language models (e.g., ESM, Evo)
- Models trained on diverse omics data for general-purpose use
- Models explicitly positioned as "foundation models for biology"
Use this category for models that truly span multiple biological domains. If a model is primarily focused on ONE domain (e.g., only proteins), use the specific category instead.
---
**8. "Biology research agents / desk research agents"**
Scope: Agentic AI systems that automate parts of the biological research workflow, particularly in silico / desk research.
Includes:
- Literature review and synthesis agents
- Hypothesis generation systems
- Experimental planning agents
- Tool/API orchestration for biological queries
- Multi-step reasoning over biological knowledge bases
- LLM wrappers with biology-specific prompting and tool use
- Governance or risk analysis agents for biology AI
IMPORTANT: This is a HIGH-PRIORITY category. Include systems that wrap existing LLMs (GPT-4, Claude, etc.) with biology-specific capabilities.
---
If a model spans multiple categories, choose the PRIMARY function emphasized in the paper. Use "General-purpose biology AI" only for truly multi-domain foundation models, not as a default.
=========================================================================
MODEL NAME RULES
=========================================================================
- `model_name` MUST be the canonical/umbrella name of the model/system introduced by the paper (the name used in the Title/Abstract, or in a sentence like "we introduce X" / "we present X").
- Do NOT use:
- a sub-component name, internal module name, or a task/agent string (e.g., "Llama-3.1-8B ... Agent ...")
- a single checkpoint/size variant as the only name when the paper clearly presents a larger named family/system.
- If the paper explicitly defines multiple official variants (e.g., "ModelX-Small", "ModelX-Large"), extract EACH as its own entry, but keep the naming consistent with the paper's official variant names.
- Preserve the paper's exact spelling/capitalization/hyphenation.
- If multiple names/aliases exist, prefer the most formal/full name used for introduction.
- If the model is UNNAMED (e.g., "our method", "the proposed network"), return exactly: `"[Unnamed model]"`
- NEVER return empty string for `model_name`. Use "[Unnamed model]" if no name is given.
=========================================================================
SAFEGUARD DEFINITIONS
=========================================================================
Safeguards are CONCRETE ACTIONS taken to reduce misuse risk or biological hazards.
Do NOT confuse safeguards with:
- Model capabilities or performance
- Standard ML practices (data cleaning, benchmarking)
- Opinions, justifications, or beliefs about risk
CRITICAL: Acknowledging risk is NOT the same as implementing a safeguard.
A paper may say "we considered the risks" or "benefits outweigh risks" without actually DOING anything to mitigate those risks. Such statements are NOT safeguards.
---
**STEP 1: Did the authors acknowledge misuse potential?**
Determine whether the paper explicitly identifies misuse potential, dual-use concerns, or biosecurity risks.
- `risk_acknowledged`: "Yes" if the paper explicitly mentions biosecurity risks, misuse, dual-use concerns, or potential for harm. "No" otherwise.
- `risk_acknowledgment_quotes`: List of verbatim quotes where risk is acknowledged, or empty list.
This is INDEPENDENT of whether they implemented safeguards. Capture acknowledgment even if no action was taken.
---
**STEP 2: Identify specific safeguards**
A safeguard must involve a CONCRETE ACTION or IMPLEMENTED MEASURE -- not just a statement or opinion.
For each safeguard, record the type and supporting quotes that describe the ACTION taken.
---
**1. "Training data curation"**
Definition: Explicit removal or filtering of hazardous data from training datasets for SAFETY reasons.
What COUNTS (concrete actions):
- "We excluded all viral genomes from Select Agents..."
- "Sequences from dangerous pathogens were removed..."
- "We filtered out gain-of-function experimental protocols..."
What does NOT count:
- Standard data cleaning (deduplication, quality filtering)
- Removing incomplete or low-quality sequences for ML purposes
- Copyright/licensing exclusions
- Statements like "we considered removing X" without confirming removal
---
**2. "Pre-release evaluation"**
Definition: Testing specifically for misuse potential or biosecurity risks BEFORE release.
What COUNTS (concrete actions):
- "We conducted red-teaming to test if the model could generate pathogenic sequences..."
- "External biosecurity experts evaluated the model's misuse potential..."
- "We tested whether the model could be prompted to produce dangerous outputs..."
What does NOT count:
- Standard accuracy, perplexity, or performance benchmarks
- Validation on held-out test sets
- General safety claims without describing specific tests
For this type, also specify `internal_or_external`:
- "Internal": Authors or their organization performed the evaluation
- "External": Third party (biosecurity org, external researchers, government)
- "Both": Both internal and external evaluations described
- "Unspecified": Not clear from the paper
---
**3. "Pre-release risk assessment"**
Definition: FORMAL threat modeling, documented risk analysis, or expert consultation BEFORE deciding to release.
What COUNTS (concrete actions):
- "We conducted a formal risk assessment in consultation with [specific experts/org]..."
- "Following the framework of [X], we analyzed potential misuse scenarios..."
- "We convened an ethics review board to evaluate release risks..."
- "Based on our threat model, we delayed release pending additional review..."
What does NOT count:
- Justification statements like "we believe benefits outweigh risks"
- Opinions like "we consider the risks to be minimal"
- Post-hoc explanations for why they released
- Vague statements like "we carefully considered safety"
- Stating that risks exist without describing what was DONE about them
- Statements like "we are looking to develop a safe and responsible offering..." without a described assessment/review procedure.
IMPORTANT: If the only "risk assessment" is a sentence like "we find the benefit of accessibility to outweigh any theoretical risks" -- this is a JUSTIFICATION, not a safeguard. Return "None described" in such cases.
---
**4. "Post-training safety techniques"**
Definition: Technical measures implemented to prevent the generation of hazardous content.
What COUNTS (concrete actions):
- "We fine-tuned the model to refuse requests for pathogen sequences..."
- "An output classifier blocks generation of [specific hazard]..."
- "The API includes a safety system prompt that prevents..."
- "User inputs are screened for hazardous intent before processing..."
What does NOT count:
- Standard output formatting
- Rate limiting for operational reasons
- General claims like "we implemented safety measures" without specifics
---
**5. "None described"**
Use this when the paper does NOT describe any concrete safeguard actions.
IMPORTANT:
- Statements of intent/values are NOT safeguards. Example: "we aim to be safe and responsible" or "we consulted on biosecurity" without describing an implemented measure.
- In such cases: set `safeguards: [{"type": "None described", "quotes": [<the intent quote(s)>], "internal_or_external": ""}]`. This preserves the evidence in quotes while correctly indicating no concrete safeguard was implemented.
Example pattern that should return "None described":
- Paper says: "We recognize dual-use potential but believe open release maximizes benefits."
- This is risk acknowledgment + justification, NOT a safeguard.
- Correct output: `risk_acknowledged: "Yes"`, `safeguards: [{"type": "None described", ...}]`
---
**Output format for safeguards:**
Each safeguard is an object with:
- `quotes`: List of verbatim quotes supporting this safeguard
- `internal_or_external`: If the type is "Pre-release evaluation", choose "Internal", "External", "Both", or "Unspecified". For ALL other types, return "".
- `type`: One of the 5 types above
=========================================================================
OTHER EXTRACTION RULES
=========================================================================
IMPORTANT: Do NOT use "N/A" for missing values. Instead:
- For string fields: use empty string ""
- For array fields: use empty array []
Only populate a field if the paper explicitly provides that information.
For EACH extracted value, you must also provide a `*_quotes` field containing a LIST of SHORT VERBATIM QUOTES from the paper that justify the extraction. If no quotes are available, use an empty list [].
QUOTE RELEVANCE: Every quote in a `*_quotes` field must DIRECTLY support the corresponding extracted value. Do not include tangentially related or thematically similar quotes. If you cannot find a directly relevant quote, use [].
---
**Parameters**
- **parameters**: If the paper explicitly states the parameter count (e.g., "7B parameters", "650M params"), extract the value as stated. Leave empty string "" if not explicitly stated. Do NOT estimate.
- **parameters_quotes**: List of verbatim quotes about model size, layers, dimensions, architecture details, etc. Include even if `parameters` is empty--these quotes enable manual estimation.
---
**Training Compute**
- **training_compute_flops**: If the paper explicitly reports training FLOPs (e.g., "1.2e23 FLOPs", "10<sup>21</sup> FLOP"), extract the value exactly as stated. Leave empty string "" if FLOPs are not explicitly reported. Do NOT estimate or calculate.
- **training_compute_quotes**: List of verbatim quotes about compute, hardware, training duration, GPUs, TPUs, cluster size, etc. Include even if `training_compute_flops` is empty--these quotes enable manual FLOP estimation.
NOTE: For agents/wrappers that orchestrate existing models without training, training compute typically does not apply. Do NOT include inference runtime (e.g., "the system runs for 12 hours") -- only include quotes about actual model training.
---
**Training Datasets**
- **training_datasets**: List of standardized dataset names used for training.
- Use canonical names like: "PDB (Protein Data Bank)", "UniRef", "UniProt", "CELLxGENE", "Human Cell Atlas", "ZINC", "ChEMBL", "PubChem", "GenBank", "RNAcentral", "AlphaFoldDB", etc.
- Return as an array of strings. Use [] (empty array) if not described.
- **training_datasets_quotes**: List of verbatim quotes describing the training data sources.
---
**Accessibility**
- **accessibility**: Accessibility status of model/weights/data. Return as an ARRAY with one or more of:
- "Open source" (training code is publicly released under an open license or as a public repo/package)
- "Open weights" (model weights/checkpoints/parameters are publicly downloadable)
- "Open data" (training data is publicly released)
- "API access" (model is available via API)
- "Web interface" (model is available via web interface)
- "Unreleased" (explicitly stated as not publicly available)
Use [] (empty array) if not described. A model can have multiple (e.g., ["Open source", "Open weights", "Open data"]).
EVIDENCE GATE: Every value you include MUST be supported by an explicit quote in `accessibility_quotes`. If a quote mentions BOTH code and weights being released (e.g., "We release our code and model weights..."), you MUST include BOTH "Open source" AND "Open weights". Do NOT default to "Unreleased" without explicit evidence--use [] instead.
- **accessibility_quotes**: List of verbatim quotes about availability.
---
**Notability**
Extract evidence of notable authorship or prestigious publication venue.
- **notability_criteria**: ARRAY of zero or more of:
- "Notable creators"
- "Prominent publisher"
Use [] if neither applies.
- **notability_criteria_quotes**: Verbatim quote(s) that directly justify the included criteria. Use [] if none apply.
QUOTE RULE: A quote must literally contain the organization name or the venue/journal name that justifies the criterion. Do NOT infer notability without an explicit quote.
**"Prominent publisher" rule**
Include "Prominent publisher" ONLY if:
1. The journal/venue name is explicitly written in the PDF (e.g., header, footer, first page), AND
2. It matches one of these venues (case-insensitive):
Nature, Nature Methods, Nature Biotechnology, Nature Machine Intelligence,
Nature Communications, Nature Chemical Biology, Nature Structural & Molecular Biology,
Cell, Cell Systems, Cell Reports, Molecular Cell,
Science, Science Advances,
PNAS, eLife
If the venue is not explicitly stated in the PDF, do NOT include "Prominent publisher".
**"Notable creators" rule**
Include "Notable creators" ONLY if:
1. The PDF contains an explicit affiliation/organization string matching one of these (case-insensitive, substring match):
Google DeepMind, DeepMind, OpenAI, Anthropic, Meta AI, FAIR, xAI, Apple,
Profluent, Chai Discovery, FutureHouse, Arc Institute, Broad Institute,
Institute for Protein Design, Baker Lab, Howard Hughes Medical Institute,
Harvard University, Stanford University, Massachusetts Institute of Technology, MIT,
University of California, Berkeley, UC Berkeley, University of Oxford, University of Cambridge
2. You include a quote containing that org name in `notability_criteria_quotes`.
Do NOT infer "Notable creators" from author names alone -- only from explicit affiliation text.
---
**Finetune Relationships**
- **finetune_of**: If this model is a fine-tune of another model (weights were modified/trained starting from a base), provide the name of the base model (e.g., "ESM-2", "RFDiffusion"). Leave empty string "" if trained from scratch or not a fine-tune.
- **finetune_of_quotes**: List of verbatim quotes identifying the base model used for fine-tuning.
- **finetune_compute_flops**: If this is a fine-tune AND the paper explicitly reports FLOPs used for fine-tuning, extract the value. Leave empty string "" if not explicitly stated or not applicable.
- **finetune_compute_quotes**: List of verbatim quotes about fine-tuning compute, hardware, duration, etc. Use [] if not applicable or not described.
---
**Uses/Implements Relationships (only for agents)**
- **uses_models**: If this is an agent or wrapper that USES other models via API or orchestration (without modifying their weights), list the models used. Return as array, e.g., ["GPT-5", "Claude 4.6 Sonnet"]. Use [] if not applicable.
IMPORTANT:
- Extract SPECIFIC model names (e.g., "GPT-4", "Claude 3.5 Sonnet", "Gemini 1.5 Pro"), NOT generic terms like "LLMs", "language models", or "foundation models". If the paper only says "we use LLMs" without specifying which model, use [].
- Only include EXTERNAL models the system calls, not internal sub-components or agents built by the same authors.
NOTE: This is DIFFERENT from `finetune_of`. Use this field for agents that CALL external models at inference time. Use `finetune_of` for models whose weights were actually trained/modified from a base.
Examples:
- PaperQA2 uses GPT-4 -> `uses_models: ["GPT-4"]`, `finetune_of: ""`
- ProGen3 fine-tuned from ProGen2 -> `finetune_of: "ProGen2"`, `uses_models: []`
- Agent says "we use LLMs" without naming them -> `uses_models: []`
- **uses_models_quotes**: List of verbatim quotes describing which models are used/called.
---
**Paper metadata: authors and organization**
- **authors**: Comma-separated list of all authors, in order. (No quotes needed.)
- **organization**: Semicolon-separated list of unique institutions from affiliations. Prefer specific lab/institute names (e.g., "Baker Lab", "Arc Institute", "Broad Institute") over generic university names when both are present. (No quotes needed.)
=========================================================================
OUTPUT FORMAT (JSON ONLY)
=========================================================================
Return exactly this structure:
{
"paper_metadata": {
"authors": "Author A, Author B, Author C, ...",
"organization": "Institution 1; Institution 2; ..."
},
"models": [
{
"model_name": "Name or [Unnamed model]",
"category_quotes": ["quote1", "quote2"],
"category_reasoning": "1-2 sentences explaining why this category was chosen.",
"category": "One of the 8 categories listed above",
"parameters_quotes": ["quote1"],
"parameters": "7B" or "",
"training_compute_quotes": ["quote1", "quote2"],
"training_compute_flops": "1.2e23" or "",
"training_datasets_quotes": ["quote1", "quote2"],
"training_datasets": ["Dataset1", "Dataset2"] or [],
"accessibility_quotes": ["quote1"],
"accessibility": ["Open source", "Open weights"] or [],
"notability_criteria_quotes": ["quote1"] or [],
"notability_criteria": ["Notable creators", "Prominent publisher"] or [],
"finetune_of_quotes": ["quote1"] or [],
"finetune_of": "ESM-2" or "",
"finetune_compute_quotes": ["quote1"] or [],
"finetune_compute_flops": "1e21" or "",
"uses_models_quotes": ["quote1"] or [],
"uses_models": ["GPT-4", "Claude 3"] or [],
"risk_acknowledgment_quotes": ["quote1"] or [],
"risk_acknowledged": "Yes" or "No",
"safeguards": [
{
"quotes": ["quote1", "quote2"],
"internal_or_external": "Internal" | "External" | "Both" | "Unspecified" | "",
"type": "Training data curation | Pre-release evaluation | Pre-release risk assessment | Post-training safety techniques | None described"
}
]
}
]
}
IMPORTANT:
- `models` is an ARRAY. Include one object per distinct model.
- All `*_quotes` fields are ARRAYS of strings.
- `category_quotes` comes BEFORE `category_reasoning` and `category` (quotes -> reasoning -> value).
- Each quote must be an EXACT verbatim quote from the paper. Do NOT paraphrase.
- `training_datasets` is an ARRAY of standardized dataset names (or [] if not described).
- `safeguards` is an ARRAY of objects.
- For `internal_or_external`: only populate for "Pre-release evaluation" type; use "" for all other types.
- If risk is acknowledged but no safeguards implemented, set `risk_acknowledged: "Yes"` AND `safeguards: [{"type": "None described", "quotes": [], "internal_or_external": ""}]`.
- Use "" (empty string) instead of "N/A" for missing string values.
- Use [] (empty array) instead of ["N/A"] for missing array values.
- `notability_criteria` must be supported by explicit quotes containing the org/venue name; if no matching quote exists, use [].
=========================================================================
PAPER
=========================================================================
Analyze the attached PDF document.
Safeguards extraction
You are provided with a research paper (attached PDF) that has ALREADY passed the relevance gate.
Your task: extract PAPER-LEVEL risk acknowledgment and safeguards information from this paper.
=========================================================================
PAPER METADATA (TITLE / DOI / AUTHORS / ORGANIZATION)
=========================================================================
Extract and return:
- `doi`: The paper DOI if present anywhere in the PDF. If not found, return "N/A".
- `title`: The paper title as written in the PDF. If not found, return "N/A".
- `authors`: Comma-separated list of all authors in order. If not found, return "N/A".
- `organization`: Semicolon-separated list of unique institutions from affiliations. If not found, return "N/A".
=========================================================================
SAFEGUARD DEFINITIONS
=========================================================================
Safeguards are CONCRETE ACTIONS taken to reduce misuse risk or biological hazards.
Do NOT confuse safeguards with:
- Model capabilities or performance
- Standard ML practices (data cleaning, benchmarking)
- Opinions, justifications, or beliefs about risk
CRITICAL: Acknowledging risk is NOT the same as implementing a safeguard.
A paper may say "we considered the risks" or "benefits outweigh risks" without actually DOING anything to mitigate those risks. Such statements are NOT safeguards.
---
**STEP 1: Did the authors acknowledge misuse potential?**
Determine whether the paper explicitly identifies misuse potential, dual-use concerns, or biosecurity risks.
- `risk_acknowledged`: "Yes" if the paper explicitly mentions biosecurity risks, misuse, dual-use concerns, or potential for harm. "No" otherwise.
- `risk_acknowledgment_quotes`: List of verbatim quotes where risk is acknowledged, or empty list.
This is INDEPENDENT of whether they implemented safeguards. Capture acknowledgment even if no action was taken.
---
**STEP 2: Identify specific safeguards**
A safeguard must involve a CONCRETE ACTION or IMPLEMENTED MEASURE—not just a statement or opinion.
For each safeguard, record the type and supporting quotes that describe the ACTION taken.
---
**1. "Training data curation"**
Definition: Explicit removal or filtering of hazardous data from training datasets for SAFETY reasons.
What COUNTS (concrete actions):
- "We excluded all viral genomes from Select Agents..."
- "Sequences from dangerous pathogens were removed..."
- "We filtered out gain-of-function experimental protocols..."
What does NOT count:
- Standard data cleaning (deduplication, quality filtering)
- Removing incomplete or low-quality sequences for ML purposes
- Copyright/licensing exclusions
- Statements like "we considered removing X" without confirming removal
---
**2. "Pre-release evaluation"**
Definition: Testing specifically for misuse potential or biosecurity risks BEFORE release.
What COUNTS (concrete actions):
- "We conducted red-teaming to test if the model could generate pathogenic sequences..."
- "External biosecurity experts evaluated the model's misuse potential..."
- "We tested whether the model could be prompted to produce dangerous outputs..."
What does NOT count:
- Standard accuracy, perplexity, or performance benchmarks
- Validation on held-out test sets
- General safety claims without describing specific tests
For this type, also specify `internal_or_external`:
- "Internal": Authors or their organization performed the evaluation
- "External": Third party (biosecurity org, external researchers, government)
- "Both": Both internal and external evaluations described
- "Unspecified": Not clear from the paper
---
**3. "Pre-release risk assessment"**
Definition: FORMAL threat modeling, documented risk analysis, or expert consultation BEFORE deciding to release.
What COUNTS (concrete actions):
- "We conducted a formal risk assessment in consultation with [specific experts/org]..."
- "Following the framework of [X], we analyzed potential misuse scenarios..."
- "We convened an ethics review board to evaluate release risks..."
- "Based on our threat model, we delayed release pending additional review..."
What does NOT count:
- Justification statements like "we believe benefits outweigh risks"
- Opinions like "we consider the risks to be minimal"
- Post-hoc explanations for why they released
- Vague statements like "we carefully considered safety"
- Stating that risks exist without describing what was DONE about them
IMPORTANT: If the only "risk assessment" is a sentence like "we find the benefit of accessibility to outweigh any theoretical risks" — this is a JUSTIFICATION, not a safeguard. Return "None described" in such cases.
---
**4. "Post-training safety techniques"**
Definition: Technical measures implemented to prevent the generation of hazardous content.
What COUNTS (concrete actions):
- "We fine-tuned the model to refuse requests for pathogen sequences..."
- "An output classifier blocks generation of [specific hazard]..."
- "The API includes a safety system prompt that prevents..."
- "User inputs are screened for hazardous intent before processing..."
What does NOT count:
- Standard output formatting
- Rate limiting for operational reasons
- General claims like "we implemented safety measures" without specifics
---
**5. "None described"**
Use this when the paper does NOT describe any concrete safeguard actions.
IMPORTANT: If a paper acknowledges risks but only provides justifications or opinions (not actions), use "None described".
Example pattern that should return "None described":
- Paper says: "We recognize dual-use potential but believe open release maximizes benefits."
- This is risk acknowledgment + justification, NOT a safeguard.
- Correct output: `risk_acknowledged: "Yes"`, `safeguards: [{"type": "None described", ...}]`
---
**Output format for safeguards:**
Each safeguard is an object with:
- `quotes`: List of verbatim quotes supporting this safeguard
- `internal_or_external`: If the type is "Pre-release evaluation", choose "Internal", "External", "Both", or "Unspecified". For ALL other types, return "N/A".
- `type`: One of the 5 types above
=========================================================================
OUTPUT FORMAT (JSON ONLY)
=========================================================================
Return exactly this structure:
{
"paper_metadata": {
"doi": "10.xxxx/xxxxx or N/A",
"title": "Full paper title or N/A",
"authors": "Author A, Author B, Author C, ... or N/A",
"organization": "Institution 1; Institution 2; ... or N/A"
},
"risk_acknowledgment_quotes": ["quote1"] or [],
"risk_acknowledged": "Yes" or "No",
"safeguards": [
{
"quotes": ["quote1", "quote2"],
"internal_or_external": "Internal | External | Both | Unspecified | N/A",
"type": "Training data curation | Pre-release evaluation | Pre-release risk assessment | Post-training safety techniques | None described"
}
]
}
IMPORTANT:
- Quotes must be EXACT verbatim quotes from the paper. Do NOT paraphrase.
- `safeguards` is an ARRAY of objects.
- If no safeguards are described, return exactly:
[{"type": "None described", "quotes": [], "internal_or_external": "N/A"}]
=========================================================================
PAPER
=========================================================================
Analyze the attached PDF document.
Paper category tags extraction
You are provided with a research paper (attached PDF) that has ALREADY passed the relevance gate.
Your task: classify the paper's biology-AI system(s) using:
- ONE `main_category` (best single fit), AND
- multiple `category_tags` (all other categories that meaningfully apply).
This is PAPER-LEVEL categorization: consider all models/systems described in the paper together.
=========================================================================
PAPER METADATA (TITLE / DOI)
=========================================================================
Extract and return:
- `doi`: The paper DOI if present anywhere in the PDF. If not found, return "N/A".
- `title`: The paper title as written in the PDF. If not found, return "N/A".
=========================================================================
CATEGORY DEFINITIONS
=========================================================================
Use the following categories (same definitions as Stage 2):
**1. "Protein engineering tools"**
Scope: AI systems for the design, optimization, prediction, or analysis of amino acid sequences and protein structures, when the purpose is to enable or drive sequence/structure engineering.
Includes:
- De novo protein design (e.g., generative models for novel proteins)
- Protein structure prediction models (e.g., AlphaFold-like models)
- Protein function prediction for engineering purposes
- Antibody design and optimization
- Enzyme engineering and optimization
- Protein-protein interaction prediction for design
- Protein fitness landscape modeling
- Directed evolution guidance models
Does NOT include: Models that only interpret experimental protein data (e.g., mass spec analysis) without a design component.
**2. "Small biomolecule design tools"**
Scope: AI systems for the optimization or de novo design of small molecules and peptides, especially for drug discovery or metabolite design.
Includes:
- Molecular generation models (VAEs, diffusion, autoregressive)
- Molecular property prediction for optimization
- Drug-target interaction prediction
- Lead optimization models
- Retrosynthesis planning
- Peptide design (short sequences, typically <50 amino acids)
- ADMET property prediction when used for design
Does NOT include: Simple QSAR models used only for screening without generative capability.
**3. "Viral vector design tools"**
Scope: AI systems that design or optimize viral vectors and capsids for gene therapy, delivery, or related applications.
Includes:
- AAV capsid design and optimization
- Viral vector tropism prediction
- Capsid engineering for improved delivery
- Lentiviral vector optimization
**4. "Genetic modification and genome design tools"**
Scope: AI systems that propose or optimize DNA/RNA sequences, edits, or larger genome designs.
Includes:
- CRISPR guide RNA design and optimization
- Gene circuit design
- Regulatory element (promoter/enhancer) design
- Codon optimization
- Synthetic genome design
- Operon and construct design
- Gene expression prediction for design purposes
- Variant effect prediction when used to guide editing
Does NOT include: Models that only interpret sequencing data without design intent.
**5. "Immune system modelling and vaccine design tools"**
Scope: AI systems for designing vaccines, immunogens, epitopes, or modeling immune responses to guide design.
Includes:
- Epitope prediction and design
- Immunogen optimization
- Vaccine antigen design
- Antibody repertoire modeling
- MHC binding prediction for vaccine design
- Neoantigen prediction for cancer vaccines
**6. "Pathogen property prediction and host-pathogen interaction tools"**
Scope: AI systems modeling pathogen traits or host-pathogen interactions, particularly for biosecurity-relevant applications.
Includes:
- Pathogen virulence prediction
- Transmissibility prediction
- Drug resistance prediction
- Immune escape / antigenic drift prediction
- Zoonotic risk assessment
- Pandemic potential modeling
- Host-pathogen protein interaction prediction
IMPORTANT: This is a HIGH-PRIORITY category for biosecurity tracking. Do not miss models in this category.
**7. "General-purpose biology AI"**
Scope: Large foundation models trained on substantial biological data that support multiple downstream tasks, or are explicitly described as "foundation", "general", or "platform" models.
Includes:
- Multi-modal biological foundation models (e.g., protein + DNA + molecules)
- Large-scale biological language models (e.g., ESM, Evo)
- Models trained on diverse omics data for general-purpose use
- Models explicitly positioned as "foundation models for biology"
Use this category for models that truly span multiple biological domains. If a model is primarily focused on ONE domain (e.g., only proteins), use the specific category instead.
**8. "Biology research agents / desk research agents"**
Scope: Agentic AI systems that automate parts of the biological research workflow, particularly in silico / desk research.
Includes:
- Literature review and synthesis agents
- Hypothesis generation systems
- Experimental planning agents
- Tool/API orchestration for biological queries
- Multi-step reasoning over biological knowledge bases
- LLM wrappers with biology-specific prompting and tool use
- Governance or risk analysis agents for biology AI
IMPORTANT: This is a HIGH-PRIORITY category. Include systems that wrap existing LLMs (GPT-4, Claude, etc.) with biology-specific capabilities.
=========================================================================
MAIN CATEGORY vs TAGS (DECISION RULES)
=========================================================================
- `main_category`: choose the single BEST-FIT category for the overall paper. If multiple categories appear, choose the PRIMARY function emphasized in the paper.
- `category_tags`: include EVERY category that meaningfully applies to any substantial model/system described in the paper.
- Tags should be a subset of the 8 categories.
- Always include `main_category` in `category_tags`.
- If the paper truly spans multiple biological domains as a foundation model/platform, use main_category="General-purpose biology AI".
Evidence requirements:
- Provide `category_quotes` as verbatim quotes that support the chosen categories/tags.
- Do NOT paraphrase quotes.
=========================================================================
OUTPUT FORMAT (JSON ONLY)
=========================================================================
Return exactly this structure:
{
"paper_metadata": {
"doi": "10.xxxx/xxxxx or N/A",
"title": "Full paper title or N/A"
},
"category_quotes": ["quote1", "quote2"] or [],
"category_reasoning": "1-3 sentences explaining the main_category and any tags.",
"main_category": "One of the 8 categories above",
"category_tags": ["Category A", "Category B"]
}
IMPORTANT:
- `category_tags` must be a non-empty array.
- `category_tags` must contain only the exact category strings listed above.
- Include `main_category` in `category_tags`.
- Quotes must be EXACT verbatim.
=========================================================================
PAPER
=========================================================================
Analyze the attached PDF document.
Notes
About the authors



Related work